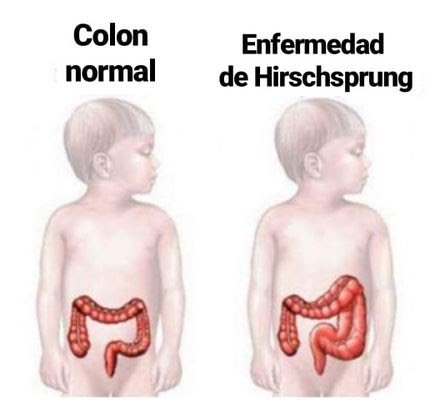
Es
una enfermedad establecida desde el nacimiento (congénito) de un bebé, con
deficiencia en la transmisión de movimientos del intestino grueso (colon), que
limita la eliminación de su contenido en forma natural y causa acumulación
progresiva del material retenido que, al paso del tiempo, deforma el volumen
intestinal generando incremento de sus dimensiones (megacolon).
En forma histórica y aún en algunas comunidades donde no se conoce esta enfermedad, se caracteriza por ausencia o escasez de evacuaciones en los primeros días del recién nacido, seguido por llanto frecuente e intenso, con aumento del volumen abdominal importante, vómitos, rechazo oral, mal estado general con rápido deterioro, capaz de producir la muerte en los primeros meses de vida.
De forma natural todos los recién nacidos tienen acumulación en sus intestinos de un material conocido como meconio, que esta formado como producto de desecho, del: líquido (amniótico) que estuvieron tomando dentro del vientre materno, combinado con secreciones y células descamadas intestinales. Este material se elimina cuando la activación nerviosa especial inducida en la fase de tranquilidad neonatal (colinérgica -por la abundancia de este neurotransmisor del sistema nervioso-), estimula los movimientos intestinales, por el envío de impulsos en dos vías (una en la capa muscular y otra por debajo de la capa mucosa), favoreciendo la eliminación del material intestinal retenido en un periodo variable de 14 a 20 hrs (promedio habitual 18hr).
Esta enfermedad está condicionada por la ausencia de tejido especializado de conducción nervioso intestinal (ganglios nerviosos) en la parte más lejana del intestino grueso (segmento recto del colon) con extensión variable hacia regiones superiores de ese intestino, que establecen su clasificación tomando como punto de referencia la unión del recto con su parte superior (sigmoides) en tres tipos básicos de defectos: largos, cortos y ultracortos. Las zonas carentes del tejido ganglionar nervioso mantienen una condición de tipo obstructivo, por la contracción predominante ante la ausencia de la estimulación nerviosa deficiente. Así de acuerdo con la extensión de la zona afectada en consecuencia, se definen las características de las manifestaciones asociadas, siendo las manifestaciones más evidentes o notorias en las formas largas, mientras que las de tipo ultracorto, representarán dificultad para su identificación en forma temprana y puede ser motivo de estreñimiento crónico en algunos pacientes a edades mayores.
Su frecuencia varía de acuerdo con las poblaciones que se han estudiado, pero en general su frecuencia se ha establecido de un caso por cada cinco mil a diez mil nacimientos, con una tendencia a aumentar ligeramente por una sospecha mayor y un diagnóstico más precoz, que disminuye la mortalidad sin diagnóstico. El defecto de segmento corto (hasta unión rectosigmoidea) es 4 veces más frecuente en los varones, que representa hasta un 80% de todos los casos, 10% puede ser de tipo largo, 8% en forma ultracorta y el resto puede incluir afectación más lejanas. El antecedente familiar se identifica en un 7% que llega hasta un 20% cuando el defecto es una forma larga.
La transmisión de la enfermedad es compleja e implica una herencia establecida por afección de varios genes. El hallar esta afección debe inducir al médico, a buscar otras alteraciones que pueden estar asociadas con esta enfermedad y ser importantes a identificar para su manejo temprano. Su diagnóstico se efectúa en el 65% de los casos durante la etapa de recién nacido (primer mes de vida), al considerar el antecedente de ausencia de eliminación del meconio en las primeras 48 horas de vida; y en el 95% de los casos, antes de cumplir el primer año de vida.
El síntoma principal que a todo familiar y/o personal médico nos hace sospechar la enfermedad, es el estreñimiento de aparición temprana en un recién nacido a término. Esta sospecha permite actualmente hacer un diagnóstico más rápido en el neonato o en el lactante pequeño y en forma asociada poder establecer el tratamiento correspondiente. El 99% de los lactantes a término eliminan el meconio en las primeras 48 horas de vida. Los prematuros eliminan más tarde el meconio, pero esta enfermedad, es rara en ellos. Considerando que sólo un 60% de los enfermos eliminan el meconio después de las 48 horas, debemos tener un grado de desconfianza con este simple signo.
Los niños mayores del primer mes manifiestan con frecuencia datos comunes de la obstrucción intestinal, como: crecimiento o distensión abdominal además de vómitos verdosos (material biliar) y ausencia de evacuaciones. La inspección anal y el estudio radiológico en forma adicional, por el médico, permitirán orientar el cuadro en forma más habitual hacia una obstrucción mecánica, pero sin excluir a esta enfermedad de otros procesos con alteración de tipo funcional. Será el médico quien se encargue de descartar otras causas con manifestaciones similares como: malformaciones intestinales, alteraciones hormonales o trastornos especiales del metabolismo.
En ocasiones, el cuadro puede aparecer en forma diferente en los primeros días de vida, con mal estado general del bebé, fiebre, rechazo a su alimentación, sensación de náuseas y/o vómitos, acompañado de aumento progresivo abdominal (enterocolitis) después de un estreñimiento que no llamó demasiado la atención, que produce que el intestino se dilate y disminuya la cantidad de sangre a la pared intestinal, alterando la vitalidad de la superficie mucosa y agregación de bacterias, con riesgo elevado de perforación.
Las dos condiciones previas permiten sospechar el padecimiento y atenderlo en forma temprana. Si la alteración es ultracorta (sin llegar a rectosigmoides), el cuadro solo establecerá un estreñimiento moderado en los primeros días de vida con eliminación un poco tardía del meconio, lo cual hace difícil sospechar la enfermedad y su diagnóstico se retrasará al igual que su tratamiento. Estos niños muestran dificultad creciente para la eliminación de las heces, llegando a notarse con el tiempo, incremento del volumen abdominal por la acumulación de evacuaciones, que se puede extender a todo el abdomen y solo mantiene al lactante inquieto por periodos cortos, llamando la atención, que este estreñimiento está presente antes del inicio del aporte de alimentos (sólidos) diferentes a la leche y, se acompañan de nauseas o vómitos ocasionales y/o recurrentes.
Como signos a identificar en estos niños para sospechar esta enfermedad, están: el aumento de volumen de la cavidad abdominal con adelgazamiento de su pared, la presencia de una red venosa visible o congestionada sobre el abdomen, una nutrición deficiente, deficiencia en su crecimiento y falta de apetito, que puede ser confundida muy fácilmente como parasitosis, por personas sin experiencia en esta enfermedad.
La evaluación médica no debe retrasarse y menos ser motivo para empleo de remedios caseros y/o tratamientos antiparasitarios, por el riesgo de la complicación letal posible. Debe ser revisado por el pediatra, neonatólogo y en especial: el cirujano pediatra, para su revisión clínica o con estudios de imagen y de biopsias de la mucosa intestinal, para definir el cuadro y establecer su manejo.
En la última década, el desarrollo de las técnicas quirúrgicas junto con los cuidados perioperatorios ha hecho descender la mortalidad y la morbilidad de la enfermedad, aunque han aumentado los casos familiares. Tan pronto como se confirma el cuadro, debe efectuarse el tratamiento quirúrgico después de vaciar las heces acumuladas en el colon dilatado. El procedimiento varía de acuerdo con edad y condiciones del paciente, retirando la zona afectada y conectando tejido funcional al extremo de eliminación.
Es posible en más de la mitad de los casos, la existencia de complicaciones inmediatas o tardías, que establecen un pronóstico a valorar en su evolución y ameritan la evaluación periódica, logrando en su mayoría que, en la vida adulta tengan buena continencia… es importante al nacimiento del bebé, estar pendiente del tiempo de su primera evacuación.
En forma histórica y aún en algunas comunidades donde no se conoce esta enfermedad, se caracteriza por ausencia o escasez de evacuaciones en los primeros días del recién nacido, seguido por llanto frecuente e intenso, con aumento del volumen abdominal importante, vómitos, rechazo oral, mal estado general con rápido deterioro, capaz de producir la muerte en los primeros meses de vida.
De forma natural todos los recién nacidos tienen acumulación en sus intestinos de un material conocido como meconio, que esta formado como producto de desecho, del: líquido (amniótico) que estuvieron tomando dentro del vientre materno, combinado con secreciones y células descamadas intestinales. Este material se elimina cuando la activación nerviosa especial inducida en la fase de tranquilidad neonatal (colinérgica -por la abundancia de este neurotransmisor del sistema nervioso-), estimula los movimientos intestinales, por el envío de impulsos en dos vías (una en la capa muscular y otra por debajo de la capa mucosa), favoreciendo la eliminación del material intestinal retenido en un periodo variable de 14 a 20 hrs (promedio habitual 18hr).
Esta enfermedad está condicionada por la ausencia de tejido especializado de conducción nervioso intestinal (ganglios nerviosos) en la parte más lejana del intestino grueso (segmento recto del colon) con extensión variable hacia regiones superiores de ese intestino, que establecen su clasificación tomando como punto de referencia la unión del recto con su parte superior (sigmoides) en tres tipos básicos de defectos: largos, cortos y ultracortos. Las zonas carentes del tejido ganglionar nervioso mantienen una condición de tipo obstructivo, por la contracción predominante ante la ausencia de la estimulación nerviosa deficiente. Así de acuerdo con la extensión de la zona afectada en consecuencia, se definen las características de las manifestaciones asociadas, siendo las manifestaciones más evidentes o notorias en las formas largas, mientras que las de tipo ultracorto, representarán dificultad para su identificación en forma temprana y puede ser motivo de estreñimiento crónico en algunos pacientes a edades mayores.
Su frecuencia varía de acuerdo con las poblaciones que se han estudiado, pero en general su frecuencia se ha establecido de un caso por cada cinco mil a diez mil nacimientos, con una tendencia a aumentar ligeramente por una sospecha mayor y un diagnóstico más precoz, que disminuye la mortalidad sin diagnóstico. El defecto de segmento corto (hasta unión rectosigmoidea) es 4 veces más frecuente en los varones, que representa hasta un 80% de todos los casos, 10% puede ser de tipo largo, 8% en forma ultracorta y el resto puede incluir afectación más lejanas. El antecedente familiar se identifica en un 7% que llega hasta un 20% cuando el defecto es una forma larga.
La transmisión de la enfermedad es compleja e implica una herencia establecida por afección de varios genes. El hallar esta afección debe inducir al médico, a buscar otras alteraciones que pueden estar asociadas con esta enfermedad y ser importantes a identificar para su manejo temprano. Su diagnóstico se efectúa en el 65% de los casos durante la etapa de recién nacido (primer mes de vida), al considerar el antecedente de ausencia de eliminación del meconio en las primeras 48 horas de vida; y en el 95% de los casos, antes de cumplir el primer año de vida.
El síntoma principal que a todo familiar y/o personal médico nos hace sospechar la enfermedad, es el estreñimiento de aparición temprana en un recién nacido a término. Esta sospecha permite actualmente hacer un diagnóstico más rápido en el neonato o en el lactante pequeño y en forma asociada poder establecer el tratamiento correspondiente. El 99% de los lactantes a término eliminan el meconio en las primeras 48 horas de vida. Los prematuros eliminan más tarde el meconio, pero esta enfermedad, es rara en ellos. Considerando que sólo un 60% de los enfermos eliminan el meconio después de las 48 horas, debemos tener un grado de desconfianza con este simple signo.
Los niños mayores del primer mes manifiestan con frecuencia datos comunes de la obstrucción intestinal, como: crecimiento o distensión abdominal además de vómitos verdosos (material biliar) y ausencia de evacuaciones. La inspección anal y el estudio radiológico en forma adicional, por el médico, permitirán orientar el cuadro en forma más habitual hacia una obstrucción mecánica, pero sin excluir a esta enfermedad de otros procesos con alteración de tipo funcional. Será el médico quien se encargue de descartar otras causas con manifestaciones similares como: malformaciones intestinales, alteraciones hormonales o trastornos especiales del metabolismo.
En ocasiones, el cuadro puede aparecer en forma diferente en los primeros días de vida, con mal estado general del bebé, fiebre, rechazo a su alimentación, sensación de náuseas y/o vómitos, acompañado de aumento progresivo abdominal (enterocolitis) después de un estreñimiento que no llamó demasiado la atención, que produce que el intestino se dilate y disminuya la cantidad de sangre a la pared intestinal, alterando la vitalidad de la superficie mucosa y agregación de bacterias, con riesgo elevado de perforación.
Las dos condiciones previas permiten sospechar el padecimiento y atenderlo en forma temprana. Si la alteración es ultracorta (sin llegar a rectosigmoides), el cuadro solo establecerá un estreñimiento moderado en los primeros días de vida con eliminación un poco tardía del meconio, lo cual hace difícil sospechar la enfermedad y su diagnóstico se retrasará al igual que su tratamiento. Estos niños muestran dificultad creciente para la eliminación de las heces, llegando a notarse con el tiempo, incremento del volumen abdominal por la acumulación de evacuaciones, que se puede extender a todo el abdomen y solo mantiene al lactante inquieto por periodos cortos, llamando la atención, que este estreñimiento está presente antes del inicio del aporte de alimentos (sólidos) diferentes a la leche y, se acompañan de nauseas o vómitos ocasionales y/o recurrentes.
Como signos a identificar en estos niños para sospechar esta enfermedad, están: el aumento de volumen de la cavidad abdominal con adelgazamiento de su pared, la presencia de una red venosa visible o congestionada sobre el abdomen, una nutrición deficiente, deficiencia en su crecimiento y falta de apetito, que puede ser confundida muy fácilmente como parasitosis, por personas sin experiencia en esta enfermedad.
La evaluación médica no debe retrasarse y menos ser motivo para empleo de remedios caseros y/o tratamientos antiparasitarios, por el riesgo de la complicación letal posible. Debe ser revisado por el pediatra, neonatólogo y en especial: el cirujano pediatra, para su revisión clínica o con estudios de imagen y de biopsias de la mucosa intestinal, para definir el cuadro y establecer su manejo.
En la última década, el desarrollo de las técnicas quirúrgicas junto con los cuidados perioperatorios ha hecho descender la mortalidad y la morbilidad de la enfermedad, aunque han aumentado los casos familiares. Tan pronto como se confirma el cuadro, debe efectuarse el tratamiento quirúrgico después de vaciar las heces acumuladas en el colon dilatado. El procedimiento varía de acuerdo con edad y condiciones del paciente, retirando la zona afectada y conectando tejido funcional al extremo de eliminación.
Es posible en más de la mitad de los casos, la existencia de complicaciones inmediatas o tardías, que establecen un pronóstico a valorar en su evolución y ameritan la evaluación periódica, logrando en su mayoría que, en la vida adulta tengan buena continencia… es importante al nacimiento del bebé, estar pendiente del tiempo de su primera evacuación.